판코니 빈혈
"오늘의AI위키"의 AI를 통해 더욱 풍부하고 폭넓은 지식 경험을 누리세요.
1. 개요
판코니 빈혈(FA)은 주로 상염색체 열성 유전자 질환으로, DNA 복구와 관련된 21개의 유전자 변이로 인해 발생한다. FA는 백혈구, 적혈구, 혈소판 발달 실패로 면역 기능 저하, 산소 전달 능력 감소, 혈전 형성 능력 저하를 유발하며, 골수 부전, 급성 골수성 백혈병(AML), 고형 종양 및 발달 이상을 특징으로 한다. 치료는 안드로겐과 조혈 성장 인자를 사용하며, 조혈모 세포 이식이 영구적인 치료법으로 사용된다. FA 환자는 AML 발병 위험이 높고, 많은 경우 성인이 되기 전에 사망한다.

더 읽어볼만한 페이지
![판코니 빈혈은 [[상염색체 열성]] 유전 패턴을 가짐.](https://cdn.onul.works/wiki/noimage.png)
2. 유전적 특징
판코니 빈혈(FA)은 대부분 상염색체 열성 유전자를 통해 유전되지만, 일부는 X 염색체와 관련되어 있는 유전 질환이다. 이 질환은 DNA 복구에 관여하는 유전자들의 변이로 인해 발생한다.
아슈케나지 유대인 집단에서는 보인자 빈도가 약 90명 중 1명으로 나타난다.[8] 이러한 이유로, 판코니 빈혈 유전자 보인자 가능성이 있는 가족에게는 유전 상담과 유전자 검사가 권장된다.
혈액학적 구성 요소인 백혈구, 적혈구, 혈소판의 발달 실패는 신체의 면역 체계, 산소 전달, 혈전 형성 능력을 감소시킨다.
2. 1. 유전 방식
판코니 빈혈(FA)은 주로 상염색체 열성 유전자 질환이다. 이는 질병을 유발하려면 두 개의 변이된 대립 유전자(각 부모로부터 하나씩)가 필요하다는 것을 의미한다. 부모 양쪽 모두 보인자일 경우, 자녀가 FA를 가질 확률은 25%이다. 약 2%의 FA 사례는 X 염색체 연관 열성으로, 어머니가 한 개의 변이된 판코니 빈혈 대립 유전자를 하나의 X 염색체에 가지고 있다면, 남성 자손이 판코니 빈혈을 나타낼 확률은 50%이다.과학자들은 21개의 FA 또는 FA 유사 유전자를 확인했다: ''FANCA'', ''FANCB'', ''FANCC'', ''FANCD1''(BRCA2), ''FANCD2'', ''FANCE'', ''FANCF'', ''FANCG'', ''FANCI'', ''FANCJ''(BRIP1), ''FANCL'', ''FANCM'', ''FANCN''(PALB2), ''FANCO''(RAD51C), ''FANCP''(SLX4), ''FANCQ''(XPF), FANCS(BRCA1), FANCT (UBE2T), FANCU(XRCC2), FANCV(REV7), 및 ''FANCW (RFWD3)''. ''FANCB''는 FA가 상염색체 열성이라는 것의 예외인데, 이 유전자가 X 염색체에 있기 때문이다. 이 유전자들은 DNA 복구에 관여한다.
아슈케나지 유대인 집단의 보인자 빈도는 약 90명 중 1명이다.[8] 판코니 빈혈의 유전자 보인자일 수 있는 가족에게는 유전 상담과 유전자 검사가 권장된다.

2. 2. 관련 유전자
판코니 빈혈(FA)은 주로 상염색체 열성 유전자 질환이다. 이는 질병을 유발하려면 두 개의 변이된 대립 유전자(각 부모로부터 하나씩)가 필요하다는 것을 의미한다. 각 자녀가 FA를 가질 확률은 25%이다. 약 2%의 FA 사례는 X 염색체 연관 열성으로, 어머니가 한 개의 변이된 판코니 빈혈 대립 유전자를 하나의 X 염색체에 가지고 있다면, 남성 자손이 판코니 빈혈을 나타낼 확률은 50%이다.과학자들은 다음과 같이 21개의 FA 또는 FA 유사 유전자를 확인했다.
판코니 빈혈(FA) 환자들은 다양한 신체적 이상을 겪을 수 있으며, 혈액학적 이상은 이 질환의 가장 심각한 임상 양상이다. FA 환자의 약 80%는 20세까지 골수 부전이 발생한다. 혈액학적 문제의 첫 징후는 보통 점상 출혈과 멍이며, 이후에 창백한 외모, 피로감, 감염이 나타난다. 거대 적혈구증이 일반적으로 혈소판 감소증보다 먼저 나타나기 때문에, FA와 관련된 전형적인 선천성 기형이 있는 환자는 증가된 적혈구 평균 적혈구 용적에 대해 평가해야 한다.[7]
''FANCB''는 FA가 상염색체 열성이라는 것의 예외인데, 이 유전자가 X 염색체에 있기 때문이다. 이 유전자들은 DNA 복구에 관여한다.
아슈케나지 유대인 집단의 보인자 빈도는 약 90명 중 1명이다.[8] 판코니 빈혈의 유전자 보인자일 수 있는 가족에게는 유전 상담과 유전자 검사가 권장된다.
3. 임상 양상
3. 1. 신체적 특징
판코니 빈혈(FA)은 골수 부전, AML, 고형 종양 및 발달 이상이 특징이다. 전형적인 특징으로는 엄지손가락 기형, 요골 결손, 작은 키, 카페오레 반점을 포함한 피부 과다 색소 침착, 비정상적인 얼굴 특징(삼각형 얼굴, 소두증), 신장 이상, 생식 능력 감소 등이 있다. 많은 FA 환자(약 30%)는 전형적인 신체 소견이 없지만, 디에폭시부탄 염색체 취약성 검사에서 염색체 파괴가 증가하면 진단을 내릴 수 있다.[6]
소안구증 및 소두증은 FA 환자에게서 흔한 선천적 결함이다.
3. 2. 혈액학적 이상
임상적으로 혈액학적 이상은 판코니 빈혈 환자에게 가장 심각한 증상이다. 40세까지 판코니 빈혈 환자의 98%는 어떤 형태의 혈액학적 이상을 보이게 된다.[6] 하지만, 몇몇 고령 환자들은 혈액학적 이상이 나타나기 전에 사망하기도 한다. 증상은 점진적으로 나타나며, 종종 완전한 골수 부전으로 이어진다. 출생 시에는 혈구 수치가 보통 정상이지만, 비정상적으로 큰 적혈구로 정의되는 거대 적혈구증/거대 적아구성 빈혈이 처음 발견되는 이상이며, 흔히 생후 10년 이내에 나타난다(발병 중앙 연령은 7세).[7] 그 후 10년 이내에 혈액학적 이상을 보이는 환자의 50% 이상이 두 개 이상의 혈구 계통의 이상으로 정의되는 범혈구 감소증을 겪게 된다. 이는 적혈구에만 영향을 미치는 다이아몬드-블랙팬 빈혈 및 주로 호중구 감소증을 유발하는 슈와흐만-다이아몬드 증후군과는 대조적이다.
가장 흔하게는 혈소판 감소증(혈소판 감소증)이 호중구 감소증(호중구 감소증)보다 먼저 나타나며, 두 증상 모두 비교적 비슷한 빈도로 나타난다. 이러한 결핍은 각각 출혈과 재발성 감염의 위험을 증가시킨다.
판코니 빈혈이 DNA 복구, 특히 상동 재조합에 영향을 미치는 것으로 알려져 있고,[1] 골수 내의 역동적인 세포 분열에 대한 현재의 지식을 고려할 때, 환자는 결과적으로 골수 부전, 골수형성이상 증후군, 급성 골수성 백혈병(AML)을 겪을 가능성이 더 높다.
골수형성이상 증후군(MDS)은 이전에는 백혈병 전 단계로 알려졌으며, 급성 골수성 백혈병(AML)과 많은 형태학적 특징을 공유하지만 몇 가지 중요한 차이점이 있는 골수 종양 질환군이다. 첫째, 미분화 전구 세포인 모세포의 비율이 항상 20% 미만이며, AML의 경우보다 이형성증이 훨씬 더 많다. 이형성증은 적혈구, 과립구, 거핵세포 전구 세포의 세포질 및 핵 형태학적 변화로 정의된다. 이러한 변화는 지연된 세포 자멸사 또는 프로그래밍된 세포사멸 실패를 반영한다. 치료하지 않으면 MDS는 약 30%의 경우에서 AML로 이어질 수 있다. FA 병리의 특성상, MDS 진단은 골수의 세포 유전학적 분석만으로는 내릴 수 없다. 실제로 골수 세포의 형태학적 분석이 수행될 때에만 MDS 진단을 확인할 수 있다. 검사 결과, MDS가 발생한 FA 환자는 MDS 이전 또는 이후에 나타나는 많은 클론 변이를 보인다. 또한 세포는 염색체 이상을 보이는데, 가장 흔한 것은 단염색체증 7과 3q 염색체의 부분적인 삼염색체증이다. 골수에서 단염색체증 7 관찰은 AML 발생 위험 증가와 매우 불량한 예후, 일반적으로 2년 이내 사망(신속한 동종 조혈모세포 이식이 옵션이 아닌 경우)과 밀접한 관련이 있다.[9]
판코니 빈혈 환자는 골수에서 20% 이상 또는 혈액에서 5~20%의 골수아세포가 존재하는 것으로 정의되는 급성 골수성 백혈병(AML) 발병 위험이 높다. 급성 전골수구 백혈병을 제외한 모든 AML 아형이 판코니 빈혈에서 발생할 수 있다. 그러나 골수단구세포 백혈병과 급성 단핵구 백혈병이 가장 흔하게 관찰되는 아형이다. 많은 골수형성이상증후군(MDS) 환자의 질병은 오래 생존할 경우 AML로 발전한다. 또한, AML 발병 위험은 골수 부전의 발병과 함께 증가한다.
20세 이전에 MDS 또는 AML이 발병할 위험은 27%에 불과하지만, 30세까지는 43%, 40세까지는 52%로 증가한다. 역사적으로 골수 이식을 하더라도 MDS/AML로 진단받은 판코니 빈혈 환자의 약 1/4이 2년 이내에 MDS/AML 관련 원인으로 사망했다.[10] 하지만 최근 발표된 증거에 따르면, 판코니 빈혈을 앓는 소아의 경우 더 이른 시기에 동종 조혈모세포 이식을 받는 것이 시간이 지남에 따라 더 나은 결과를 가져오고 있다.[11]
판코니 빈혈과 관련된 마지막 주요 혈액학적 합병증은 골수 부전으로, 이는 부적절한 혈구 생성을 의미한다. 판코니 빈혈 환자에게서 여러 유형의 부전이 관찰되며, 일반적으로 골수형성이상증후군 및 급성 골수성 백혈병보다 먼저 발생한다. 혈구 감소의 감지는 일반적으로 치료의 필요성과 이식 가능성을 평가하는 데 사용되는 첫 번째 징후이다. 대부분의 판코니 빈혈 환자는 초기에는 안드로겐 요법과 조혈 성장 인자에 반응하지만, 이는 특히 세포유전학적 이상이 있는 환자에게서 백혈병을 촉진하는 것으로 나타났으며, 간 선종 및 선암종을 포함한 심각한 부작용을 동반한다. 남은 유일한 치료법은 골수 이식이지만, 공여자가 관련 없는 경우(5년 생존율 30%) 판코니 빈혈 환자에서는 성공률이 비교적 낮다. 따라서 HLA가 일치하는 형제자매로부터 이식하는 것이 필수적이다. 또한 판코니 빈혈 환자는 염색체 손상에 대한 감수성이 증가하므로, 이식 전 처치에 고용량의 방사선이나 면역억제제를 사용할 수 없으며, 이로 인해 환자가 이식편대숙주병을 앓을 위험이 증가한다. 모든 예방 조치를 취하고 골수 이식을 생후 10년 이내에 수행하면 2년 생존 확률이 89%까지 높아질 수 있다. 그러나 이식을 10세 이상에 수행하면 2년 생존율이 54%로 떨어진다.
Zhang 등의 최근 보고서에 따르면 FANCC-/- 세포에서 골수 부전의 메커니즘을 조사했다.[12] 그들은 고산소 혈액과 저산소 골수 조직 사이를 이동하는 조혈 세포 및 전구 세포에서 보이는 것과 같은 저산소증-재산소화의 지속적인 사이클이 조기 세포 노화로 이어지고, 따라서 조혈 기능의 억제를 초래한다고 가설을 세우고 성공적으로 이를 입증했다. 노화는 세포 자멸사와 함께 골수 부전에서 발생하는 조혈 세포 고갈의 주요 메커니즘을 구성할 수 있다.
4. 분자 병리 기전
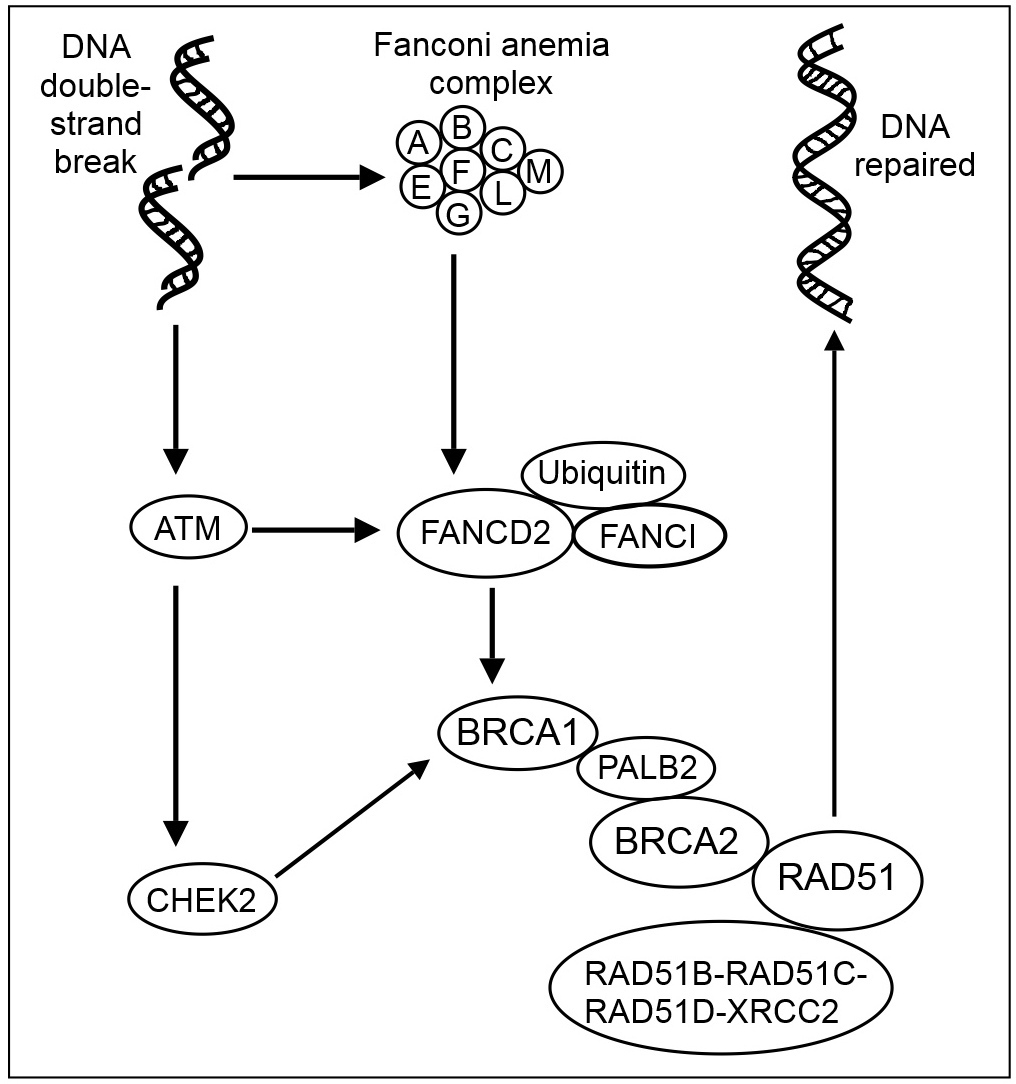
DNA 이중 가닥 손상은 ATM 단백질 키나아제를 활성화하고, 판코니 빈혈 코어 복합체 (FANCA/B/C/E/F/G/L/M)를 활성화한다.[13] FA 코어 복합체는 FANCD2와 FANCI를 유비퀴틴화한다.[14] ATM은 CHEK2와 FANCD2를 인산화하여 활성화한다.[15] CHEK2는 BRCA1을 인산화한다.[16] 유비퀴틴화된 FANCD2는 BRCA1 및 RAD51과 복합체를 이룬다.[17]
PALB2 단백질은 DNA 이중 가닥 절단 부위에 BRCA1, BRCA2 및 RAD51을 모으고, RAD51 paralog 복합체 RAD51B-RAD51C-RAD51D-XRCC2 (BCDX2)의 구성원인 RAD51C에도 결합한다. BCDX2 복합체는 손상 부위에서 RAD51 모집 또는 안정화에 관여한다.[19] RAD51은 DNA의 상동 재조합 복구에 중요한 역할을 한다. 이 과정에서 ATP 의존적 DNA 가닥 교환이 일어나 단일 가닥이 상동 DNA 분자의 염기쌍을 이룬 가닥을 침범한다. RAD51은 이 과정의 상동성 검색 및 가닥 쌍 형성 단계에 관여한다.
판코니 빈혈(FA) 관련 유전자는 총 22개이며,[20][21] 이들은 손상된 DNA 인식 및 복구에 관여한다. 유전적 결함은 DNA 복구 능력을 저해한다.
다음은 FA 단백질 중 일부에 대한 표이다.
사람의 경우 FANC 유전자의 돌연변이 결함을 가진 개인에게서 불임이 발견된다.[40] 쥐의 경우, 정원 세포, 전 렙토텐기 정모 세포 및 렙토텐기, 접합기 및 초기 태사기의 감수 분열 단계에 있는 정모 세포에 FANC 단백질이 풍부하게 존재한다.[40] 이는 FANC 단백질에 의해 매개되는 재조합 복구 과정이 생식 세포 발달, 특히 감수 분열 동안 활성화되며, 이러한 활성의 결함이 불임으로 이어질 수 있음을 시사한다.
4. 1. 판코니 빈혈 코어 복합체
판코니 빈혈(FA)을 유발하는 유전자는 22개가 있으며,[20][21] 그중 하나는 유방암 감수성 유전자 BRCA2이다. 이들은 손상된 DNA의 인식 및 복구에 관여한다. 유전적 결함이 있으면 DNA를 복구할 수 없게 된다. 8개의 단백질로 구성된 FA 코어 복합체는 손상으로 인해 DNA 복제가 중단될 때 일반적으로 활성화된다. 코어 복합체는 DNA를 복구하기 위해 다른 클러스터에서 BRCA2와 결합하는 작은 단백질인 유비퀴틴을 추가한다. 이 과정이 끝나면 유비퀴틴이 제거된다.[2]최근 연구에 따르면 FANCA, -B, -C, -E, -F, -G, -L 및 -M의 8가지 단백질이 핵에서 코어 단백질 복합체를 형성하는 것으로 나타났다. 현재 모델에 따르면, 이 복합체는 FANCA 및 FANCE의 핵 국소화 신호에 따라 세포질에서 핵으로 이동한다. 조립은 복제 스트레스, 특히 DNA 손상에 의해 활성화된다. 이는 DNA 가교제(마이토마이신 C 또는 시스플라틴 등) 또는 FANCM 단백질에 의해 감지되는 활성 산소종(ROS)에 의해 발생한다.[22]
조립 후, 단백질 코어 복합체는 FANCL 단백질을 활성화하며, 이는 E3 유비퀴틴 연결 효소로 작용하여 FANCD2[23][24][25][26] 및 FANCI를 모노유비퀴틴화한다.[27][28]
FANCD2-L로도 알려진 모노유비퀴틴화된 FANCD2는 BRCA1/BRCA2 복합체와 상호 작용한다. 자세한 내용은 알려져 있지 않지만, 유사한 복합체는 게놈 감시에 관여하고 DNA 복구 및 염색체 안정성에 관련된 다양한 단백질과 관련되어 있다.[29][30] 복합체 내의 FA 단백질에 치명적인 돌연변이가 있으면, 시스플라틴, 디에폭시부탄[31] 및 마이토마이신 C와 같은 가교제에 의해 발생하는 손상에 대한 반응에서 볼 수 있듯이, DNA 복구가 훨씬 덜 효과적이다. 골수는 이러한 결함에 특히 민감하다.
이온화 방사선에 반응하는 또 다른 경로에서, FANCD2는 이중 가닥 DNA 절단에 의해 활성화된 단백질 복합체 ATM/ATR에 의해 인산화되어 S기 체크포인트 조절에 참여하는 것으로 생각된다. 이러한 경로는 FA-D1 또는 FA-D2 환자에서 S기 체크포인트의 결함의 특징인 방사선 저항성 DNA 복제의 존재로 입증되었다. 이러한 결함은 세포의 제어할 수 없는 복제를 쉽게 유발하며, 이러한 환자에서 급성 골수성 백혈병(AML)의 빈도가 증가하는 것을 설명할 수도 있다.
4. 2. 기타 기능
FA 단백질은 DNA 복구 외에도 자가포식 및 리보솜 생합성에 관여한다.[21] FANCC, FANCA, FANCF, FANCL, FANCD2, BRCA1 및 BRCA2는 손상된 미토콘드리아를 제거하는 과정인 미토파지에 필요하다.[32][33][34][35][36] BRCA1(FANCS라고도 함)은 리보솜 DNA (rDNA) 프로모터 및 종결자와 핵소체에서 상호 작용하며, 핵소체는 리보솜 생합성이 시작되는 세포 위치이며 rDNA의 전사에 필요하다.[37] FANCI는 대형 리보솜 서브유닛의 생산, 프리리보솜 RNA(pre-rRNA)의 처리, RNAPI에 의한 pre-rRNA의 전사, 성숙한 28S 리보솜 RNA (rRNA) 수준 유지, 리보솜에 의한 단백질의 전반적인 세포 번역에 관여한다.[20] 핵소체에서 FANCI는 주로 유비퀴틴화되지 않은 형태이다.[20] FANCA는 정상적인 핵소체 형태를 유지하고, pre-rRNA의 전사 및 전반적인 세포 번역을 위해 필요하다.[38] FANCC, FANCD2, FANCG는 또한 정상적인 핵소체 형태를 유지하는 데 필요하며 FANCG는 전반적인 세포 번역에 필요하다.[38]리보솜 생합성 또는 단백질 번역에서 핵소체 외부 FA 단백질의 역할이 있을 수 있는데, FANCI와 FANCD2가 폴리솜과 관련된 유일한 FA 단백질이기 때문이다.[38] 디스케라토시스 선천성, 다이아몬드-블랙팬 빈혈, 슈바흐만-다이아몬드 증후군을 포함한 다른 유전성 골수 부전 증후군도 리보솜 생합성 또는 단백질 번역에 결함이 있으며, 이러한 다른 질병과 마찬가지로 FA도 리보솜병일 수 있다.[20][21][39]
사람의 경우, 불임은 FANC 유전자의 돌연변이 결함을 가진 개인의 특징 중 하나이다.[40] 쥐의 경우, 정원 세포, 전 렙토텐기 정모 세포 및 렙토텐기, 접합기 및 초기 태사기의 감수 분열 단계에 있는 정모 세포에 FANC 단백질이 풍부하게 존재한다.[40] 이는 FANC 단백질에 의해 매개되는 재조합 복구 과정이 생식 세포 발달, 특히 감수 분열 동안 활성화되며, 이러한 활성의 결함이 불임으로 이어질 수 있음을 시사한다.
FANCC는 산화 스트레스에 대한 세포 반응에서 중요한 역할을 한다. FANCC는 NADPH 시토크롬 P450 환원 효소와 상호 작용하여 활성 산소 생성을 증가시키고, 항산화 글루타치온 생성에 중요한 글루타치온-S-전이효소와 관련이 있다. Cu/Zn 슈퍼옥시드 디스뮤테이스와 FANCC 변이를 가진 쥐에서는 조혈 작용이 불완전하다. FANCC 유전자는 STAT1에 결합하여 STAT135의 수용체 결합 및 인산화를 보조하여 항암 작용을 한다. FANCC는 아폽토시스를 억제하여 세포 증식 정지와 세포 주기의 진행에 관여한다. 산화 스트레스 방어와 관련된 판코니 빈혈 단백질에는 FANCG가 있으며, CYP2E1과 상호 작용하기 때문에 시토크롬 활성 산소를 해독하는 역할을 한다. FANCG는 복제 후 수정 단백질인 XRCC9와 동일하며[66], FANCG도 그 내부의 류신 지퍼에 의해 DNA에 직접 작용할 가능성을 시사한다.
5. 치료
치료의 첫 번째 단계는 안드로겐과 조혈 성장 인자이며, 환자의 50~75%만이 반응을 보인다. 보다 영구적인 치료법은 조혈모 세포 이식이다.[42] 잠재적 기증자가 없는 경우, 수혜자의 HLA형에 일치하는 구원자 형제를 착상 전 유전자 진단을 통해 낳을 수 있다.[43][44]
6. 예후
많은 환자들이 결국 급성 골수성 백혈병(AML)으로 발전한다. 고령 환자는 두경부, 식도, 위장, 외음부 및 항문 암이 발생할 가능성이 매우 높다.[45] 성공적인 줄기 세포 이식을 받아 판코니 빈혈과 관련된 혈액 문제를 치료한 환자라도 암 징후를 관찰하기 위해 정기적인 검사를 받아야 한다. 많은 환자들이 성인이 되지 못한다.
환자의 대부분은 결국 급성 골수성 백혈병(AML)을 발병한다. 환자가 연장되면 두경부암, 식도암, 위장관암, 자궁 경부암, 항문암 발병 가능성이 매우 높아진다.[55] 골수 이식이 성공하여 판코니 빈혈에 따른 혈액 질환 문제가 해소된 환자라도, 암의 징후를 발견하기 위해 정기적인 검사가 필요하다. 많은 환자가 성인이 되기 전에 사망한다.
골수형성이상증후군(MDS)은 전백혈병으로도 알려졌던 질환으로, 일련의 골수 종양성 질환을 지칭하며, 급성 골수성 백혈병(AML)과 몇 가지 주요한 차이점을 제외하고는 많은 점에서 형태학적으로 공통점을 갖는다. 치료하지 않으면 MDS는 30%의 사례에서 AML로 이행한다. 판코니 빈혈의 병리적 특성 때문에 골수형성이상증후군의 진단은 골수의 세포유전학적 분석만으로는 이루어질 수 없다. 골수형성이상증후군의 진단은 골수 세포의 형태학적 분석이 이루어진 후에 확정된다.
판코니 빈혈에서는 급성 골수성 백혈병(AML) 발병 위험이 높아진다. AML은 골수 내 골수아세포가 20% 이상 존재하거나, 혈액 내 골수아세포가 5-20%를 차지하는 것으로 확정된다. 골수 이형성 증후군 환자 중 많은 수가 오래 생존하면 결국 급성 골수성 백혈병으로 이행된다. 게다가 진행성 급성 골수성 백혈병의 위험은 골수 부전 발병과 함께 증가한다.
골수 이형성 증후군이나 급성 골수성 백혈병 중 하나를 20세 전에 발병할 확률은 27%로 낮지만, 30세까지는 위험이 43%로 상승하며, 40세까지는 52%에 달한다. 골수 이식을 시행하더라도, MDS나 AML을 앓은 판코니 빈혈 환자의 4명 중 1명은 MDS나 AML 관련 증상으로 2년 이내에 사망한다.[56]
판코니 빈혈에 합병되는 중요한 혈액학적 합병증으로 마지막은 골수 부전이다. 이는 혈액 세포의 생산이 불충분한 것으로 정의된다. 판코니 빈혈 환자에서는 여러 유형의 부전이 나타나며, 많은 경우 골수형성이상증후군(MDS)이나 급성 골수성 백혈병(AML)보다 먼저 발생한다.
참조
[1]
학술지
The Fanconi Anemia DNA Repair Pathway: Structural and Functional Insights into a Complex Disorder
2014-04-17
[2]
학술지
Susceptibility pathways in Fanconi's anemia and breast cancer
2010-05
[3]
학술지
How high are carrier frequencies of rare recessive syndromes? Contemporary estimates for Fanconi Anemia in the United States and Israel
[4]
문서
WhoNamedIt|synd|61
[5]
학술지
Familiäre infantile perniziosaartige Anämie (perniziöses Blutbild und Konstitution)
[6]
학술지
The inherited bone marrow failure syndromes
[7]
문서
EMedicine|article|960401|Fanconi Anemia|clinical
[8]
학술지
Fanconi anemia in Ashkenazi Jews
[9]
학술지
Numerical chromosomal changes and risk of development of myelodysplastic syndrome-acute myeloid leukemia in patients with Fanconi anemia
[10]
학술지
Hematologic abnormalities in Fanconi anemia: an International Fanconi Anemia Registry study
[11]
학술지
Allogeneic hematopoietic stem cell transplantation in Fanconi anemia: the EBMT experience
[12]
학술지
Hypoxia-reoxygenation induces premature senescence in FA bone marrow hematopoietic cells
[13]
학술지
Susceptibility pathways in Fanconi's anemia and breast cancer
[14]
학술지
The Fanconi anemia protein FANCM is controlled by FANCD2 and the ATR/ATM pathways
[15]
학술지
Coordinated action of the Fanconi anemia and ataxia telangiectasia pathways in response to oxidative damage
[16]
학술지
Tumor suppressor CHK2: regulator of DNA damage response and mediator of chromosomal stability
[17]
학술지
S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51
[18]
학술지
PALB2: the hub of a network of tumor suppressors involved in DNA damage responses
[19]
학술지
Rad51 paralog complexes BCDX2 and CX3 act at different stages in the BRCA1-BRCA2-dependent homologous recombination pathway
[20]
학술지
Fanconi anemia protein FANCI functions in ribosome biogenesis
2019-02-12
[21]
학술지
Inherited bone marrow failure in the pediatric patient
2022-08-11
[22]
학술지
FANCM connects the genome instability disorders Bloom's Syndrome and Fanconi Anemia
2009-12
[23]
학술지
BRCA1-independent ubiquitination of FANCD2
[24]
학술지
Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway
[25]
학술지
BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures
http://www.genesdev.[...]
[26]
학술지
Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks
[27]
학술지
Structural and biochemical basis of interdependent FANCI-FANCD2 ubiquitination
2022-11-17
[28]
학술지
Differential functions of FANCI and FANCD2 ubiquitination stabilize ID2 complex on DNA
2020-07-03
[29]
학술지
Biallelic inactivation of BRCA2 in Fanconi anemia
[30]
학술지
Tumorigenesis and a DNA repair defect in mice with a truncating Brca2 mutation
[31]
서적
Fanconi Anemia
[32]
학술지
Fanconi Anemia Proteins Function in Mitophagy and Immunity
2016-05-05
[33]
학술지
Novel functions of Fanconi anemia proteins in selective autophagy and inflammation
2016-08-09
[34]
학술지
Impaired mitophagy in Fanconi anemia is dependent on mitochondrial fission
2016-09-06
[35]
학술지
Fanconi Anemia Genes, of Menders and Sweepers
2016-05-23
[36]
논문
Autophagy: Inflammatory pathology of Fanconi anaemia
https://pubmed.ncbi.[...]
2016-06
[37]
논문
The identification of a novel role for BRCA1 in regulating RNA polymerase I transcription
2016-10-18
[38]
논문
Fanconi anemia A protein participates in nucleolar homeostasis maintenance and ribosome biogenesis
2021-01
[39]
논문
A new frontier in Fanconi anemia: From DNA repair to ribosome biogenesis
2022-03-01
[40]
논문
Uncoupling of transcription and translation of Fanconi anemia (FANC) complex proteins during spermatogenesis
[41]
논문
Role of Fanconi DNA repair pathway in neural stem cell homeostasis
[42]
문서
Fanconi Anemia
https://emedicine.me[...]
[43]
서적
The Debate About Genetic Engineering (Ethical Debates)
https://archive.org/[...]
Rosen Central
[44]
논문
Preimplantation diagnosis for Fanconi anemia combined with HLA matching
[45]
웹사이트
"Constitutional chromosomal instability: a case with three primary and sequential cancers"
http://www.urologyjo[...]
Institut Biologia Fonamental de Barcelona
2010-04-13
[46]
웹사이트
Anemia, Fanconi
http://icmmt.alere.c[...]
[47]
간행물
Fanconi's Anemia
Orphanet
2003-10
[48]
논문
Susceptibility pathways in Fanconi's anemia and breast cancer
http://content.nejm.[...]
[49]
웹사이트
Synd 61
http://www.whonamedi[...]
[50]
논문
Familiäre, infantile perniciosähnliche Anämie (perniziöses Blutbild und Konstitution)
[51]
논문
Fanconi anemia in Ashkenazi Jews
[52]
웹사이트
Fanconi Anemia Treatment & Management: Approach Considerations, Supportive Care, Hematopoietic Stem Cell Transplantation and Androgen Therapy
https://emedicine.me[...]
2022-07-08
[53]
서적
The Debate About Genetic Engineering (Ethical Debates)
Rosen Central
[54]
논문
, Preimplantation diagnosis for Fanconi anemia combined with HLA matching
[55]
웹사이트
"Constitutional chromosomal instability: a case with three primary and sequential cancers"
http://www.urologyjo[...]
Institut Biologia Fonamental de Barcelona
2010-04-13
[56]
논문
Hematologic abnormalities in Fanconi anemia: an International Fanconi Anemia Registry study
https://pubmed.ncbi.[...]
1994-09-01
[57]
논문
Hypoxia-reoxygenation induces premature senescence in FA bone marrow hematopoietic cells
[58]
논문
FANCM connects the genome instability disorders Bloom's Syndrome and Fanconi Anemia
http://www.sciencedi[...]
[59]
논문
BRCA1-independent ubiquitination of FANCD2
http://linkinghub.el[...]
[60]
논문
Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway
http://linkinghub.el[...]
[61]
논문
BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures
http://www.genesdev.[...]
[62]
논문
Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks
http://www.sciencema[...]
[63]
논문
Biallelic inactivation of BRCA2 in Fanconi anemia
http://www.sciencema[...]
[64]
논문
Tumorigenesis and a DNA repair defect in mice with a truncating Brca2 mutation
[65]
논문
International Fanconi Anemia Registry: relation of clinical symptoms to diepoxybutane sensitivity
[66]
논문
The Fanconi anaemia group G gene FANCG is identical with XRCC9
[67]
논문
The Fanconi Anemia DNA Repair Pathway: Structural and Functional Insights into a Complex Disorder
http://www.annualrev[...]
2014-04-17
[68]
논문
Susceptibility pathways in Fanconi's anemia and breast cancer
http://content.nejm.[...]
2010-05
[69]
웹사이트
How high are carrier frequencies of rare recessive syndromes? Contemporary estimates for Fanconi Anemia in the United States and Israel
http://onlinelibrary[...]
2011-07-07
[70]
웹사이트
Synd 61
http://www.whonamedi[...]
[71]
저널
Familiäre, infantile perniciosähnliche Anämie (perniziöses Blutbild und Konstitution)
본 사이트는 AI가 위키백과와 뉴스 기사,정부 간행물,학술 논문등을 바탕으로 정보를 가공하여 제공하는 백과사전형 서비스입니다.
모든 문서는 AI에 의해 자동 생성되며, CC BY-SA 4.0 라이선스에 따라 이용할 수 있습니다.
하지만, 위키백과나 뉴스 기사 자체에 오류, 부정확한 정보, 또는 가짜 뉴스가 포함될 수 있으며, AI는 이러한 내용을 완벽하게 걸러내지 못할 수 있습니다.
따라서 제공되는 정보에 일부 오류나 편향이 있을 수 있으므로, 중요한 정보는 반드시 다른 출처를 통해 교차 검증하시기 바랍니다.
문의하기 : help@durumis.com